Genomic Data Processing Overview
The GDC uses submitted FASTQ or BAM formatted sequence and microarray data to generate derived analysis data. This includes analyses such as tumor sequence variant calls, RNA-Seq gene expression quantification values, and copy-number segmentation values.
Sequence data is aligned (or realigned) to the latest human genome reference. The resulting alignments are then processed to produce derived data. The alignment and derived data are available to users via the GDC Data Portal. Array data is processed using data type specific methods.
Each phase of processing is standardized into common pipelines that use open source sequence analysis tools. All sequence data submitted to the GDC is subjected to analysis through these standard pipelines. When data is successfully processed, it is made available through the GDC Data Portal and other access tools. If the data processing reveals underlying issues in the data, the associated files will be recalled and will not be available through the GDC.
The genomic data processing pipelines were developed in consultation with senior experts in the field of cancer genomics and are regularly evaluated and updated as current tools and parameter sets are improved and developed.
Reference Genome and Alignment Workflow
Reference genome alignment is the first step of data processing for all sequencing-based workflows. While different alignment algorithms are used for each case depending on read length and type, all alignments are performed on the same version of the GRCh38 reference genome. See the GDC Documentation site for details on the algorithm used for each pipeline. Viral and decoy sequences are included, which draw reads that would not normally map to the human genome, provide information on the presence of oncoviruses, and allow for a more accurate alignment. The current virus decoy set contains 10 types of human viruses, including human cytomegalovirus (CMV), Epstein-Barr virus (EBV), hepatitis B (HBV), hepatitis C (HCV), human immunodeficiency virus (HIV), human herpes virus 8 (HHV-8), human T-lymphotropic virus 1 (HTLV-1), Merkel cell polyomavirus (MCV), simian vacuolating virus 40 (SV40) and human papillomavirus (HPV).
An initial alignment is performed separately on each read group, which is defined as a set of reads that originates from one Illumina sequencing lane. The subsequent set of alignments that originate from a single aliquot are then merged. Pipeline-specific details about the alignment and downstream analyses can be found in their respective section or documentation site.

GDC Pipeline Overviews
Brief summaries of the workflow used by the GDC are listed below. Each summary has a link to its corresponding section of the GDC Documentation Website. The GDC Documentation website contains details about each step of the pipeline, the command-line parameters used to run each step, and information about the corresponding files available at the GDC Data Portal.
DNA-Seq WXS Somatic Variant Analysis
The DNA-Seq Somatic Variant Analysis pipeline identifies and characterizes somatic mutations by comparing reference alignments from tumor and normal samples from the same case. The validity of these mutations is assessed using internal algorithms and external variant databases. A co-cleaning step is implemented by recalibrating base quality scores and realigning indels for a more accurate alignment. Four separate algorithms (MuSE, Mutect2, Pindel, Varscan2) are then used to perform variant calling on paired tumor/normal samples to identify somatic mutations. Variants are annotated independently and with information from external databases such as dbSNP and OMIM. All annotated variant calls from one project are then aggregated into one MAF file per variant calling pipeline. MAF files are filtered to remove any potentially erroneous or germline variant calls. After they are filtered, open-access Somatic MAFs are available to the general public, whereas the unfiltered MAFs are available only to dbGaP-authorized investigators.
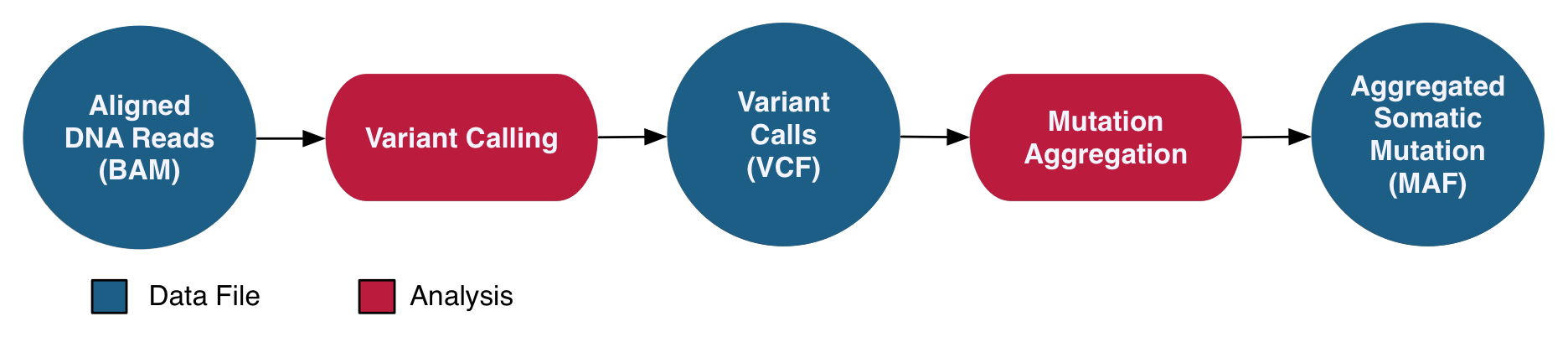
See the GDC Documentation website for an overview of the:
DNA-Seq WGS Somatic Variant Analysis
The DNA-Seq WGS data is aligned using the same method as the WXS pipeline documented above. WGS variant calling uses a pipeline developed by the Sanger Institute. This pipeline calls somatic variants using CaVEMan and Pindel, copy number variants using ASCAT-NGS, and structural variants using BRASS.
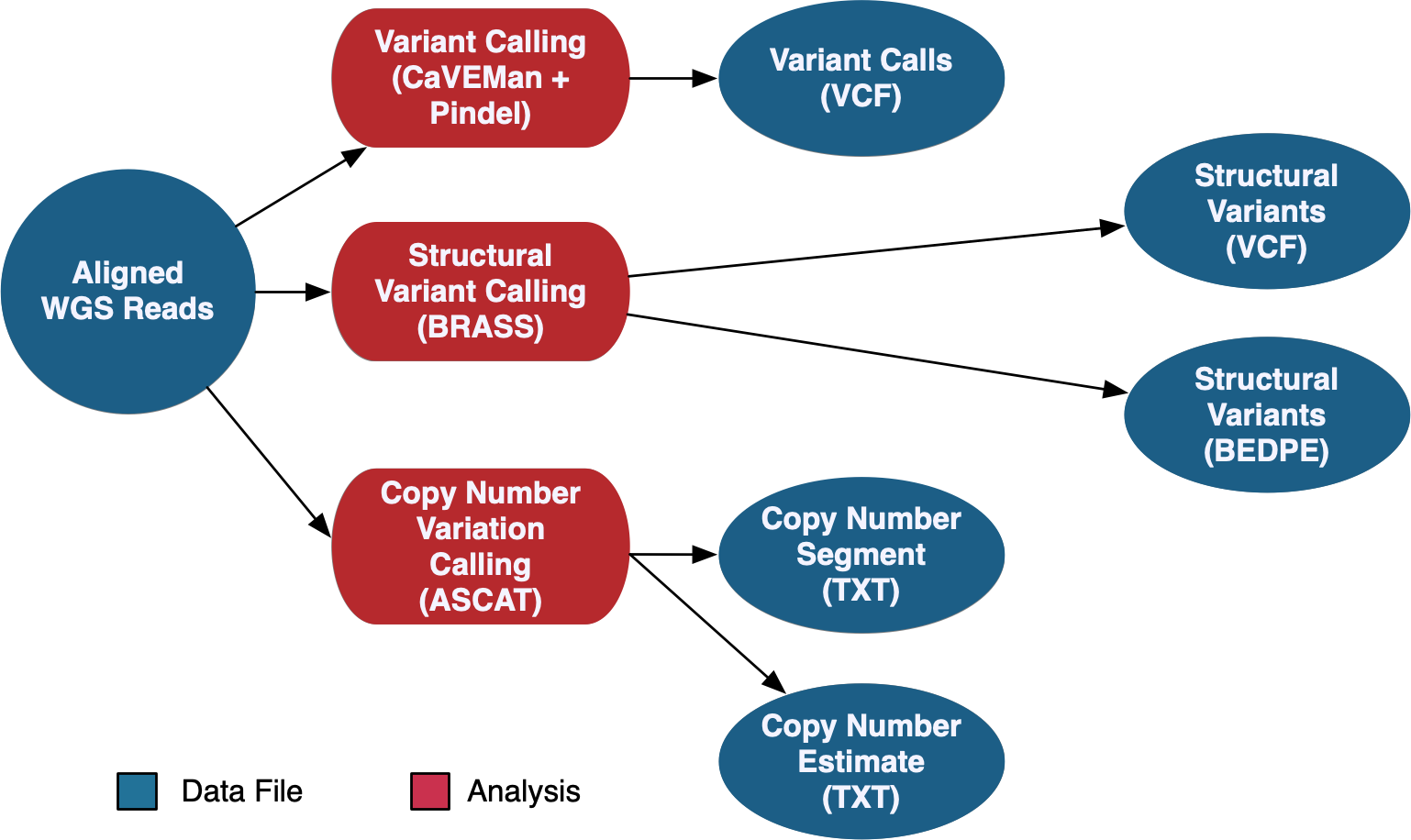
See the GDC Documentation website for an overview of the:
RNA-Seq Gene Expression Analysis
The RNA-Seq Analysis pipeline quantifies protein-coding gene expression based on the number of reads aligned to each gene. A "two-pass" method is used in which RNA-Seq reads are first aligned to the reference genome to detect splice junctions. A second alignment is then performed using the information from splice junctions to increase the quality of the alignment. Read counts are measured on a gene level using STAR and normalized using the Fragments Per Kilobase of transcript per Million mapped reads (FPKM) and FPKM Upper Quartile (FPKM-UQ) methods with custom scripts. Transcript fusion files are also generated using STAR Fusion and Arriba. HTSeq quantification is no longer supported as of Data Release 32 with the new GENCODE v36 data model.
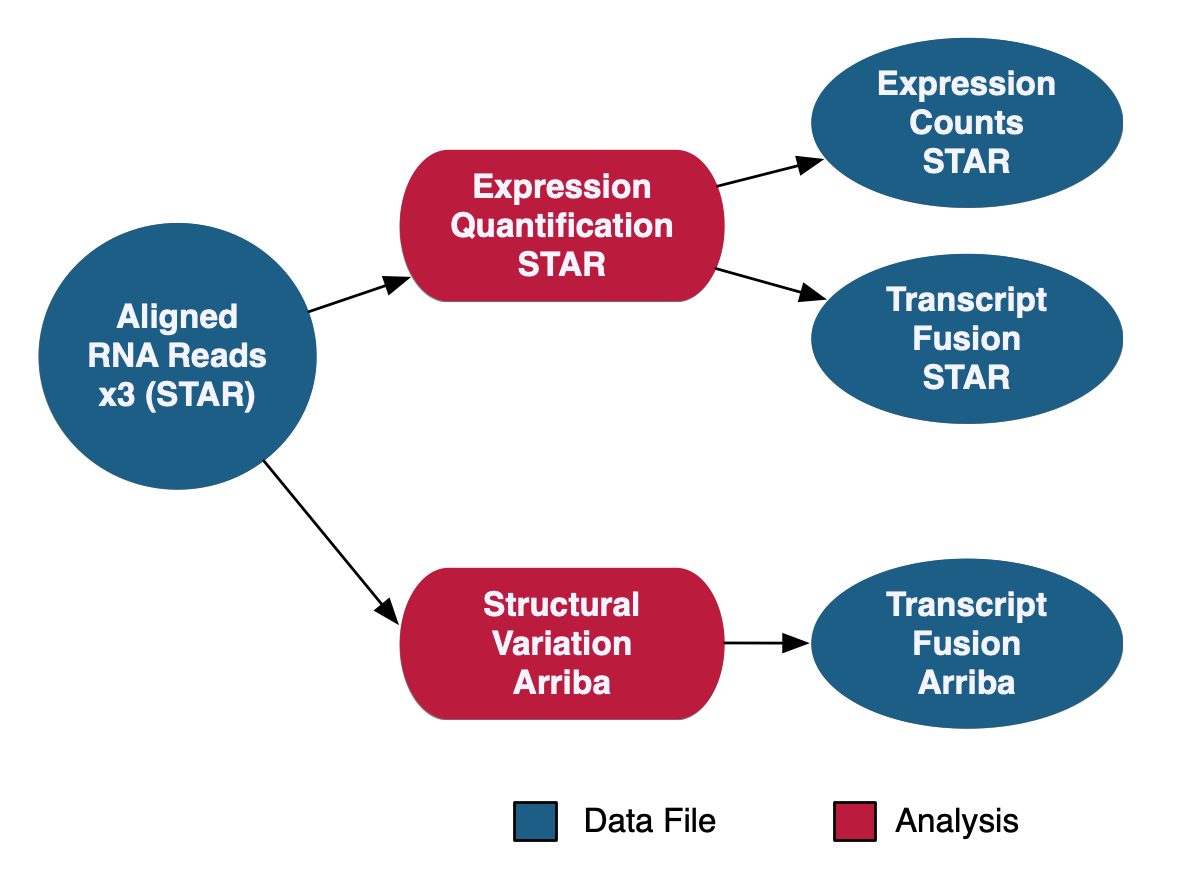
See the GDC Documentation website for a detailed overview of the:
scRNA-Seq Gene Expression Analysis
The scRNA-Seq Analysis pipeline generates counts using CellRanger, which are available in both filtered and raw format. Seurat is then used to perform a secondary expression analysis on the counts, which produces coordinates for several methods of graphical representation, differentially expressed genes, and the full analysis in loom format.
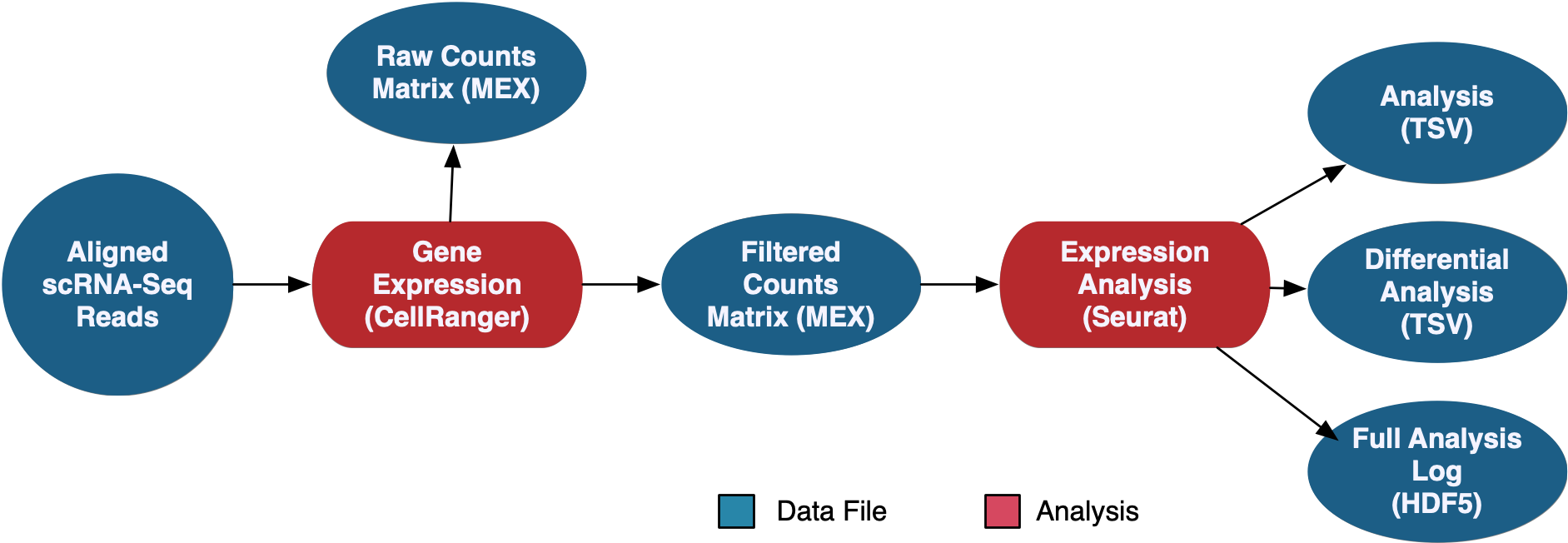
See the GDC Documentation website for a detailed overview of the:
miRNA-Seq Analysis
The miRNA-Seq pipeline quantifies micro-RNA gene expression. The names and genomic locations of each miRNA are retrieved from miRBase, and the expression levels are measured and normalized post-alignment. Normalization is performed using the Reads per Millions (RPM) method. Expression levels for known miRNAs and observed miRNA isoforms are generated for each sample.
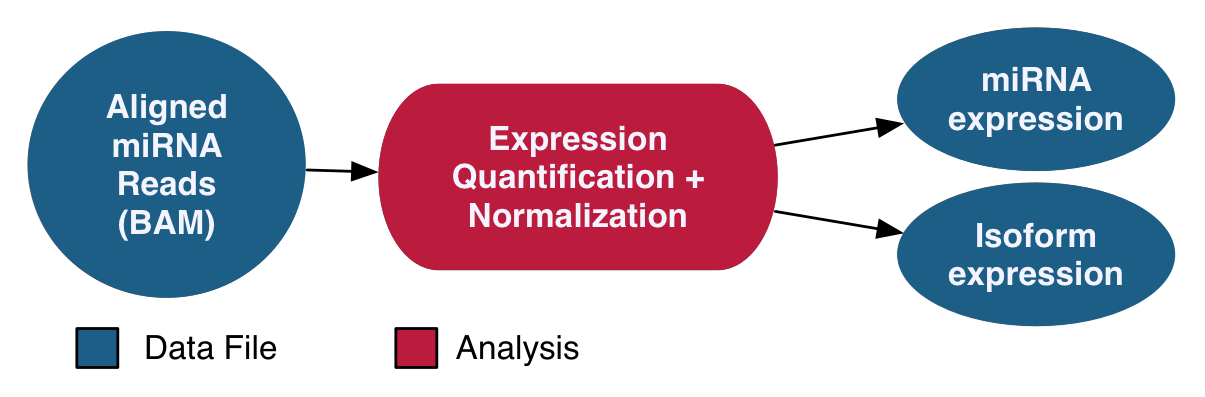
See the GDC Documentation website for a detailed overview of the:
Methylation Array Analysis
At the GDC, methylation beta values are measured at known CpG sites based on array intensities. Methylation data at the GDC is provided with methylation beta value files as well as masked methylation arrays, which have potential germline information removed. As of Data Release 32, methylation liftover files are no longer supported at GDC.
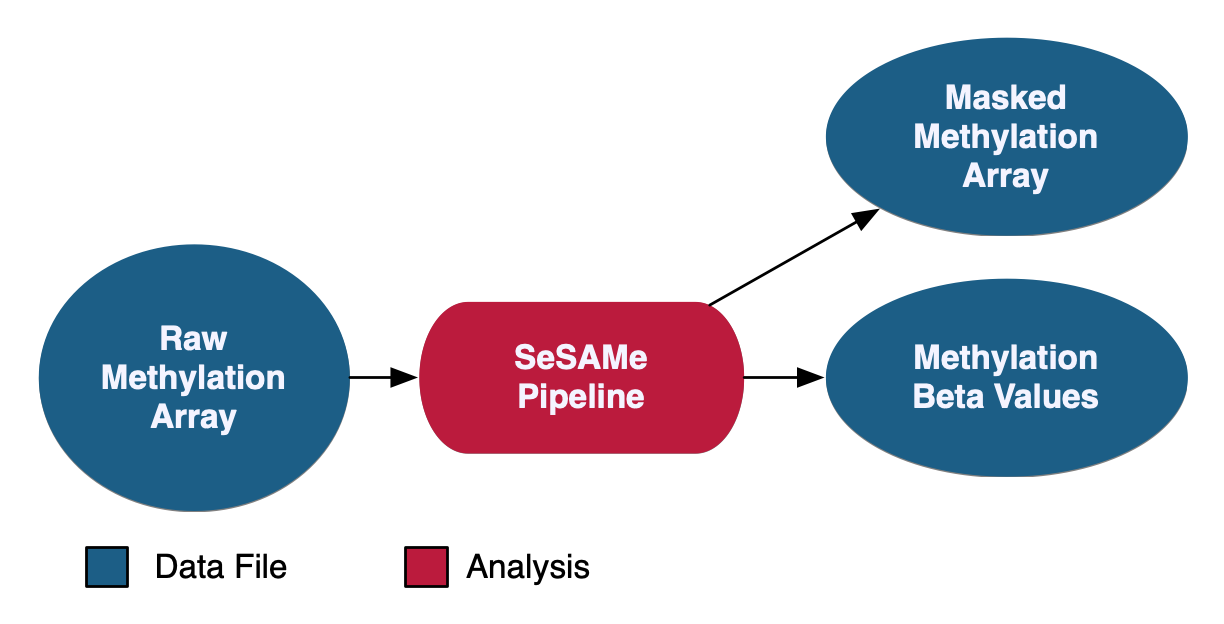
See the GDC Documentation website for a detailed overview of the:
Pipeline Implementation
GDC pipelines are packaged as a series of Docker containers. Docker can wrap up a complete environment that contains everything a bioinformatics pipeline needs to run. This includes code, runtime, tools, and, libraries. This method significantly improves reproducibility and portability of bioinformatics software in Linux systems. Realignment annotations, such as the docker ID, time cost, and exact command used in the docker container are stored as properties of the workflow for each file created. All other QC metrics and realignment tool logs are saved as individual files in the object store. For data remediation, the GDC examines all QC results manually for problem detection. The GDC will establish criteria and implement automatic remediation steps in the workflow.